
Информационно-просветительский гуманитарный проект «12 месяцев» — это цикл материалов о необычных людях – пациентах с редкими (орфанными) болезнями, о которых не написано в студенческих учебниках. Считается, что вероятность встретить на профессиональном пути редкого пациента у обычного врача ничтожно мала, поэтому в академических аудиториях им не уделяют должного внимания, что в повседневной жизни приводит к диагностическим ошибкам, упущенному времени и поломанными судьбами и жизням. Проект «12 месяцев» реализуют студенты и ординаторы – будущие и нынешние специалисты, активно изучающие генетические методы диагностики, их место в современной врачебной работе. Материалы готовятся на кафедре патологической анатомии СЗГМУ им. И.И. Мечникова (Санкт-Петербург) при поддержке научно-практического журнала «Гены и Клетки», группы компаний ИСКЧ, блога истории медицины и порталов Indicator.Ru и «Нейроновости». Каждая часть проекта состоит из двух или трех материалов: рассказа о заболевании (чаще всего с видеотаймлайном его изучения), и пациентской истории. Научные редакторы проекта — Алексей Паевский и Роман Деев. Новый цикл статей посвящен спинально-мышечной атрофии.
Знаете ли вы, что в организме человека более 600 мышц, и каждая, сокращаясь, вызывает определенное движение. Совокупность таких движений позволяет нам ходить, играть на музыкальных инструментах, и даже улыбаться. «Движение — это жизнь, а жизнь — это движение!» — так сказал Аристотель. «Движение – это навык который необходимо освоить» — так говорим мы, авторы текста. Та же ходьба представляется нам чем-то обыденным и не требует особенной сосредоточенности, но вспомните детей, которые учатся ходить. Первые шаги даются очень непросто, при этом, как только ребенок осваивает этот навык, ходьба не вызывает сложностей. Приобретенные двигательные навыки не только формируют личность человека, но и его профессию, а следовательно положение в обществе. Но задумывались ли вы о том, как именно возникает движение?
Чтобы осуществить осознанное движение, задействуется длинная цепь событий. Головной мозг является первым звеном цепи. Он формирует и составляет план движения, который передается в спинной мозг. Спинной мозг, передавая импульс по нервам, включает определенные группы мышц. Упрощенно, таким образом возникает определенное движение. Путь от головного мозга к мышцам сложен и требует слаженной работы анатомических структур, участвующих в реализации движения, и существует масса заболеваний, которые могут повлиять на звенья этой цепи. В зависимости от того, на каком этапе произошла поломка, развивается определенная клиническая картина заболевания. Так, при повреждении нервных клеток (нейронов) спинного мозга, импульс не передается на нервы, а, следовательно и на мышцы, в результате чего движения не происходит. Повреждение нейронов спинного мозга может происходить по разным причинам: от банальной травмы до сложных генетических аномалий, к последним и прикован интерес генетиков и врачей в последние годы.
Одним из наиболее часто встречаемых генетических заболеваний, при котором происходит повреждение спинного мозга, является спинальная мышечная атрофия (СМА).
СМА представляет собой генетическое заболевание, которое приводит к дегенерации нейронов спинного мозга. Их прогрессирующее разрушение приводит к мышечной слабости и гибели мышечных волокон. Проявления заболевания в конечном итоге приводят к полному обездвиживанию и нередко к летальному исходу.
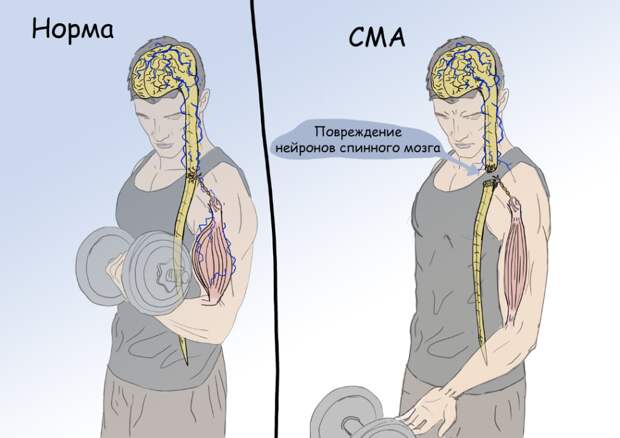
СМА является ведущей генетической причиной младенческой смертности во всем мире. Распространенность СМА крайне велика и насчитывает примерно 1 на 6000–10 000 новорожденных.
Первое научное описание заболевания произошло в 1890-х годах. Сначала Гвидо Вердниг описал тяжелую форму СМА у двух пациентов с прогрессирующей слабостью. Позднее, Иоган Гоффманн описал ещё 7 подобных случаев. Поэтому СМА иногда называют болезнью Верднига-Гоффмана. Точно так же более легкие формы СМА, описанные Кугельбергом и Веландером, иногда называют болезнью Кугельберга-Веландера.
После работ Верднига и Гоффмана, были описаны другие клинические формы, создана классификация заболевания. Но не было ясно по каким причинам погибают нейроны спинного мозга.
Одним из важнейших шагов в понимании причин развития СМА стала идентификация гена «выживаемости мотонейронов 1» (SMN1), ответственного за развитие СМА в 1995. С этого начался новый виток понимания СМА.
Стало ясно, что 95% случаев СМА вызваны мутацией гена SMN1 на длинном плече 5-й хромосомы. Он отвечает за синтез белка SMN. У человека есть две формы гена SMN: SMN1 и SMN2. Оба гена почти идентичны. Однако на основе гена SMN1 образуется полноразмерный функциональный белок SMN, в то время как ген SMN2 приводит к синтезу укороченного и нестабильного белка. Только~5–10% продуктов SMN2 будут функционально активными. Пациенты со СМА, у которых отсутствует функционирующий ген SMN1, зависят только от работы SMN2.Следовательно, они находятся в хроническом состоянии дефицита белка SMN.

Кодируемый белок, называемый SMN, встречается повсеместно в эукариотических клетках, а его полное отсутствие приводит к гибели сразу после зачатия.
Точная роль белка SMN в функционировании и развитии нейронов до конца не изучена. Причины, по которым его отсутствие оказывает такой разрушительный эффект, неизвестны.
Недавние достижения в понимании функционирования белка SMN показали, что СМА-это заболевание не только двигательных нейронов. Также существует взаимосвязь с врожденными пороками сердца и патологией чувствительных нервов.
К чему же ведет дефицит белка SMN?
При дефиците белка SMN происходит характерное нарушение работы скелетных мышц. У пациентов отмечаются нарушения произвольных движений — ползания, ходьбы, удержания головы, глотания. При этом когнитивные функции не затрагиваются, и, как сообщается, пациенты имеют интеллект от среднего до выше среднего.
Принято классифицировать СМА на несколько типов.
СМА типа 0 — самая редкая, но самая тяжелая форма. Возникает с минимальным присутствием белка SMN. Начало заболевания происходит ещё до рождения младенца, в результате чего они менее подвижны, и часто рождаются с артрогрипозом (ограниченные деформации/контрактуры суставов) и гипотонией (слабым мышечным тонусом), что приводит к смерти до или сразу после рождения ребенка. У некоторых из них имеется дыхательная недостаточность, паралич мышц лица и/или пороки сердца, приводящие к смерти в младенческом возрасте.
Тип I — наиболее распространенная форма СМА (~ 45% случаев). Она также известна как болезнь Верднига-Гоффмана и связана с тяжелой формой мышечной слабости, проявляющейся при рождении или в течение первых нескольких месяцев жизни. Большинство пациентов, обычно имеют генерализованную мышечную слабость, включая, помимо прочего, неспособность контролировать движения головы и неспособность сидеть без посторонней помощи. Из-за слабости дыхательных мышц у них затруднено дыхание и повышен риск аспирации. Младенцы испытывают трудности с глотанием и сосанием, что приводит к сложностям при кормлении.
У детей со СМА II типа, также известной как болезнь Дубовица, заболевание проявляется в более позднем детстве в возрасте от шести до 18 месяцев. Дети могут сидеть без посторонней помощи; однако они не могут стоять и ходить. Постепенно нарастает мышечная слабость, что существенно сокращает продолжительность жизни. У пациентов наблюдаются сколиоз (патологический изгиб позвоночника), тремор (непроизвольное дрожание) пальцев и слабость дыхательных мышц.
Приблизительно 30% пациентов со СМА относятся к типу III, называемому болезнью Кугельберга-Веландера, которая начинается в возрасте 18 месяцев и старше. Как правило, у пациентов развивается мышечная слабость различной степени, что приводит к гетерогенным симптомам. Хотя большинство из них могут ходить самостоятельно, у некоторых наблюдается прогрессирующая слабость мышц. Пациенты обычно испытывают трудности при подъеме по лестнице, страдают от мышечных спазмов.
Подобно III типу, у пациентов с IV типом заболевания СМА начинается относительно поздно, а именно во взрослом возрасте 30 лет и старше. Этот тип составляет менее 5% от общего числа случаев и считается легкой формой. У пациентов обычно развивается мышечная слабость, которая преимущественно поражает мышцы ног и бедер, а затем распространяется на плечи и руки. Тем не менее, они способны достигать нормального двигательного развития и имеют среднестатистическую продолжительность жизни.
В настоящее время лечение в значительной степени является паллиативным.
Заболевания легких, которые развиваются в результате слабости дыхательных мышц, у пациентов со СМА вызывают дыхательную недостаточность и являются наиболее частой причиной смерти среди больных. Для увеличения продолжительности жизни используются различные способы респираторной поддержки. Но, к сожалению, это не влияет на причину, и болезнь прогрессирует.
Из-за мышечной слабости пациенты склонны к быстрой утомляемости жевательных мышц и затруднениям глотания, а также запорам, задержке опорожнения желудка. Пациенты требуют пристального внимания к рациону питания, поскольку они более склонны к расстройствам метаболизма.
Пациенты часто страдают от ортопедических осложнений, таких как сколиоз, подвывих бедра, и более других подвержены рискам переломов. Использование физиотерапевтических процедур, таких как лечебная физкультура, массаж, растяжение мышц, необходимо для поддержания функции мышц, а применение упражнений на растяжку и пассивного движения суставов помогает избежать формирования контрактур (тугоподвижности) суставов.
Традиционные методы лечения были сосредоточены на уменьшении различных клинических проявлений СМА, но не на первопричину заболевания.
Прошло немногим более 10 лет с открытия гена, ответственного за развитие СМА, и уже в 2010 году были проведены первые доклинические исследования генной терапии. На мышиной модели было показано улучшение двигательных функций в группе, получавшей лечение. Это произвело революцию, и дало надежду тысячам больным детям и их родителям, на то, что грозное заболевание может быть излечено.
Спустя еще 6 лет тяжелой работы, в декабре 2016 был одобрен первый геннотерапевтический препарат, направленный на причину заболевания — нусинерсен.
Нусинерсен, останавливает белок, который подавляет считывание с гена SMN 2, таким образом считывание идет более активно, и увеличивается количества нормально функционирующего белка.
Эффективность Нусинерсена была продемонстрирована в клинических испытаниях у 121 пациента со СМА. Введения первой дозы проводилось детям младше 7 месяцев. В исследовании оценивался процент пациентов с улучшением моторных показателей в группах препарата и плацебо. FDA (агентство Министерства здравоохранения и социальных служб США, один из федеральных исполнительных департаментов) попросило спонсора провести промежуточный анализ, чтобы как можно раньше оценить результаты исследования. Было показано, что 40% пациентов, получавших Спинразу, достигли улучшения двигательных показателей (способность удерживать головку, сидеть без поддержки), тогда как ни у одного из пациентов контрольной группы этого не произошло.
Таким образом был преодолён барьер применения генной терапии в клинической практике. Это дало еще больший толчок развитию направления и внедрению государственных программ по снабжению пациентов необходимыми препаратами, поскольку препарат стоит 118 000 долларов США за один флакон и 708 000 долларов США за первый год лечения. В Российской Федерации зарегистрирован с 2019 года, с 2020 внесен в перечень жизненно необходимых и важнейших лекарственных препаратов.
Другим направлением в генной терапии является система доставки и внедрения в геном человека недостающего гена при помощи определенных вирусов, поскольку вирусы обладают способностью проникать в клетки, и встраивать свою генетическую информацию в генетический код зараженной клетки. Лекарство при этом называется вирусным вектором.
Вирусные векторы конструируются путём удаления патогенных генов, и их замены последовательностями, кодирующими интересующий ген. Таким образом вирусный вектор, проникнув в клетку, внедряет «недостающий» ген, с помощью которого клетка синтезирует белок.
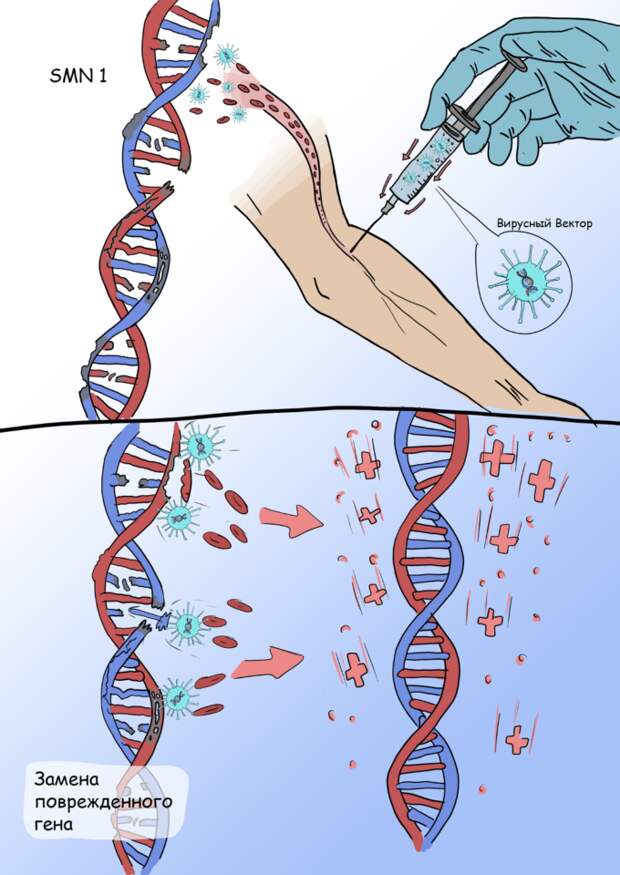
Онасемноген абепарвовек — геннотерапевтический препарат, основанный на модели вектора, ассоциированного с аденовирусом. Он осуществляет доставку гена SMN1 в клетки, что позволяет организму вырабатывать функционирующий белок SMN. Исследование фазы I/II, оценивающее безопасность и эффективность онасемногена у 15 пациентов со СМА I типа, показало, что 100% пациентов были живы через 20 месяцев по сравнению с ожидаемыми 8% при естественном течении заболевания. Кроме того, были замечены значительные двигательные улучшения: 11 из 15 участников, получавших лечение, смогли сидеть без посторонней помощи, что никогда не достигается без лечения.
Онасемноген стоит около 2,125 миллионов долларов за одну инъекцию и, как сообщается, является самым дорогим лекарством в мире, при этом терапия используется всего единожды. В Российской Федерации препарат зарегистрирован с 2021 года и пока не внесен в перечень жизненно необходимых и важнейших лекарственных препаратов.
В 2022 году Российская фармацевтическая компания BIOCAD начала разработку первого отечественного препарата генной терапии для лечения спинальной мышечной атрофии. Препарат имеет название ANB-004, и, представляет собой рекомбинантный аденоассоциированный вирус-вектор. Сейчас проходит первая фаза клинических испытаний.
Другим классом препаратов являются малые молекулы — низкомолекулярные вещества, влияющие на биологические процессы.
Представитель этой группы препарат рисдиплам — лекарство для перорального применения, который увеличивает считывание с гена SMN2 и, таким образом, увеличивает уровень функционального белка SMN. Было проведено множество испытаний фазы II/III, которые показали эффективность в отношении улучшения двигательной функции.
Эффективность рисдиплама оценивалась в двух клинических исследованиях с ранней и поздней формой СМА. В исследовании с ранним началом СМА был включен 21 пациент, средний возраст которых на момент начала исследования составлял 6,7 месяца. Эффективность устанавливалась на основе способности сидеть без поддержки не менее пяти секунд и выживаемости без постоянной вентиляции. После 12 месяцев лечения 41% пациентов были в состоянии сидеть самостоятельно более пяти секунд. После 23 месяцев лечения 81% пациентов остались живы без постоянной вентиляции легких, что является заметным улучшением по сравнению с типичным прогрессированием заболевания без лечения.
Пациентов с поздним началом СМА оценивали во втором исследовании, которое включено 180 пациентов со СМА в возрасте от 2 до 25 лет. Первичной конечной точкой было изменение общего балла MFM32 (шкала, используемая для оценки мелкой и крупной моторики у людей с неврологическими расстройствами) по сравнению с исходным уровнем на отметке в один год терапии. У пациентов, принимавших рисдиплам, наблюдалось увеличение баллов в среднем на 1,36 через год по сравнению с уменьшением на 0,19 у пациентов, получавших плацебо (неактивное лечение), что показывает эффективность препарата.
Стоимость рисдиплама — составляет около $300 тыс. в год. в России препарат зарегистрирован в декабре 2020 года, входит в перечень жизненно необходимых и важнейших лекарственных препаратов с 2020 года.
Многие исследования по препаратам продолжаются по сей день, а пациенты регулярно наблюдаются и проходят осмотры.
Несмотря на то, что результаты лечения пациентов, получавших генную терапию, кажутся более благоприятными, чем исторические исходы при СМА, у пациентов наблюдаются значительные двигательные нарушения. Генная терапия относительно новое направление в медицине, но уже видится весьма эффективным и перспективным. Как любое развивающееся направление, генная терапия будет модернизироваться, улучшаться и в дальнейшем, вероятно, будет решением огромной массы генетических заболеваний.
Современное понимание генетических заболеваний формирует задачу разработать наиболее эффективное, доступное и переносимое лекарственное средство. Эффект от лечения геннотерапевтическими агентами уже виден и связан со значимым улучшением течения заболевания. Многие научно-исследовательские и опытно-конструкторские коллективы работают над поставленной задачей — создание мира без спинальной мышечной атрофии.
Текст: Ирина Опря, Юрий Слепов, Максим Пушкин
Свежие комментарии