Сегодня в нашей исторической рубрике — совсем необычная статья. Это достаточно длинная, но захватывающая история о том, как в 1960-е годы советская ученая Татьяна Бунина под руководством знаменитого вирусолога Льва Зильбера искала «инфекционную» причину возникновения такого страшного заболевания, как боковой амиотрофический склероз. Сейчас, во время Десятилетия науки и технологий, которое особое внимание уделяет работе с опытом, накопленном наукой, такие истории поиска, удач и ошибок, очень важны для современных ученых. Этот текст написал Евгений Броновицкий, аспирант кафедры общей биологии и биоэкологии Государственного университета просвещения как научную работу, но читается это исследование на одном дыхании.
Медленные вирусные инфекции центральной нервной системы
Середина XX века – эпоха, ознаменованная плодотворной работой молекулярных биологов. Роль и структура «главной молекулы жизни» ДНК расшифрованы [13], а механизмы работы генетического аппарата обретают вполне ясные очертания[ 14]. С этого момента начинается активная работа по «разгадке» множества проблем в биологии, которые невозможно было решить из-за отсутствия понятийного аппарата в этой области науки.
Одной из таких работ стали изучения болезней почесухи овец (скрепи) и трансмиссивной губчатой энцефалопатии (куру). На сегодняшний день известно, что это трансмиссивные прионовые заболевания [15]. Однако, тогда ограниченность в знаниях давала о себе знать, на тот момент научному сообществу все еще не хватало информации для более точной идентификации природы этих заболеваний, невзирая на то, что были получены достоверные доказательства, что за инфицирующим агентом стояло нечто большее, нежели вирус, так как высеять его не удавалось [16]. При введении здоровым животным ликвора из головного мозга больных, несмотря на его обработку растворами формалина различной концентрации, длительному нагреванию [17], обработками рибо- и дезоксирибонуклеазами [18] он сохранял инфицирующую активность. Возможно, причиной такого тупика, останавливающего дальнейшие рассуждения исследователей, была главенствующая на тот момент «Центральная догма» молекулярной биологии, созданная Френсисом Криком, согласно, которой информация могла передаваться только в одном направлении – от ДНК к белку, но никак не наоборот [19]. Множество лабораторий мира было занято изучением заболеваний, поражающих центральную нервную систему, с неидентифицированным инфицирующим агентом – заболеваниями медленных вирусных инфекций (МВИ) центральной нервной системы. Концепция МВИ была предложена Бьерном Сигурдсоном в 1954 г., в результате длительных наблюдений за овцами, страдающими почесухой и заболеванием Висна-маеди. Он обнаружил схожие клинические признаки у этих заболеваний: длительный инкубационный период и абсолютная смертность. Немногим позже, исследователем был выделен РНК-содержащий вирус заболевания Висна-маеди [20]. В 1957 г. к медленным вирусным инфекциям овец добавилось заболевание индейцев каннибалов Новой Гвинеи – куру, описанное Даниелем Гайдузеком (будущим Нобелевским лауреатом), которое по своим клиническим проявлениям было схожим со скрепи и висну [21]. А в 1971 г. исландский исследователь Халдэр Тормар предложил классификацию медленных вирусных инфекций ЦНС, в которую включил и БАС [20].
Поиск возможной этиологии БАС
Гипотеза о возможной инфекционной природе БАС была предложена Л. О. Даршкевичем еще в 1911. Эта гипотеза получила широкую поддержку среди советских исследователей, которые рассматривали данное заболевание как экзогенное, инфекционно-токсическое и, вероятно, вирусное, что в дальнейшем определило вектор развития исследования природ БАС [22]
В течение почти десяти лет под руководством выдающихся советских ученных, вирусолога Льва Зильбера и невропатолога Николая Коновалова, А.М. Гардашьян и Татьяна Бунина проводили экспериментальные работы по воспроизведению БАС у Macacus rhesus путем введения экстрактов мозга умерших пациентов, страдавших различными формами БАС в ЦНС обезьян, предполагая, что инфицирующим началом заболевания должен быть вирусный агент [23].

Татьяна Бунина
Работы по изучению этиологии БАС группой Зильбера берут свое начало в результатах исследования болезни Куру, которыми занималась лаборатория медленных вирусных инфекций. В 1966 году Даниэл Гайдузек (с которым сотрудничал Зильбер) с соавторами опубликовали работу, в которой показали возможность переноса куру-подобного синдрома от человека к шимпанзе. В данном исследовании восьми шимпанзе интроцеребрально была инокулирована суспензия, приготовленная из головного мозга различных пациентов, достоверно умерших от болезни куру.

Даниэл Гайдузек
В промежутке от 18 до 21 месяцев у трех животных из восьми инокулированных начал развиваться синдром прогрессивной церебральной атаксии. Животные достигали терминальной стадии менее, чем за десять месяцев от начала дебюта заболевания. Позднее исследователям удалось перенести куру-подобный синдром и низшим приматам [24]. В множественных попытках изолировать инфицирующий агент исследователями было выделено 53 различных вируса из различных тканей шимпанзе, страдающих куру-подобным синдромом. Некоторыми штаммами из выделенных вирусов были реинокулированы нескольким шимпанзе. Однако, после смерти этих животных в тканях головного мозга не было обнаружено патоморфологических и гистологических изменений, характерных для заболевания куру [25].
Советские исследования этиологии БАС
Для проверки гипотезы вирусной природы БАС Татьяной Буниной совместно с А.Н. Гардашьян (Институт эпидемиологии и микробиологии им. Н.Ф. Гамалея АМН СССР) в 1956 году был инициирован грандиозный эксперимент на обезьянах, который продолжался более 10 лет. Для эксперимента Бунина отобрала 10 из более чем 200 пациентов, наблюдавшихся в Институте неврологии АМН СССР, страдавших наиболее выраженными формами БАС: бульбарной, шейно-грудной и крестцовой. У данных пациентов, находившихся на стационарном лечении post mortem (вскрытие проводилось не позднее 4,5 часов после смерти) была взята биопсия продолговатого и спинного мозга. Фрагменты ткани, растертые в физиологическом растворе в присутствии антибиотиков, послужили инокулирующим материалом для экспериментов на обезьянах. За все время работы было заражено 45 обезьян Macacus rhesus. В качестве контроля в эксперименте было инокулировано дополнительно 6 обезьян суспензией спинного и продолговатого мозга пациентов, погибших от травм, но без каких-либо признаков нейродегенеративного процесса [22].

Лев Зильбер
На первом этапе эксперимента команда Зильбера «заразила» только часть обезьян. Из них у 9 приматов было отмечено развитие нейродегенеративного процесса, предположительно связанного с отмиранием двигательных нейронов. А у трех животных на первом этапе была отмечена развернутая картина БАС-подобного синдрома, идентичная с таковой у пациентов, страдающих БАС, инокулятом которых иммунизировали животных. Более того, оказалось возможным выделить на определенных этапах эксперимента неврологические формы БАС, аналогичные таковым у человека [22].
Систематизация предварительных результатов эксперимента на обезьянах
После первых успешных попыток переноса заболевания от человека к приматам исследователи приняли решение расширить круг реципиентов. Следующими экспериментальными организмами стали мыши и морские свинки [23]. Однако, наблюдение за зараженными животными в течение нескольких лет не показало у них развития БАС-подобного синдрома или какого-либо иного нейродегенеративного процесса. Полученные результаты эксперимента на обезьянах лишь укрепили уверенность исследователей в вирусной природе БАС [26,27].
Для дальнейшего подтверждения своих выводов исследователями были организованы биохимические, иммунологические и гистологические исследования различных тканей и мозга, заболевших и не заболевших обезьян, которым была введена суспензия мозга от пациентов с БАС, а также изучение иммунологических характеристик сыворотки крови больных различными формами БАС людей (в том числе и семейными формами). Гардашьян была вовлечена в работу с иммунологической составляющей БАС. В течение длительного времени исследователь пыталась высеять (обнаружить) вирусный агент, который предположительно мог вызывать развитие БАС у экспериментальных животных, но ни одна из попыток не увенчалась успехом [23,28].
В результате исследования были охарактеризованы серологические составляющие заболевания БАС. Было установлено, что уровень антител, а конкретно фракции иммуноглобулинов G (IgG), возрастает на момент манифестации клинических симптомов (парезы, параличи, мышечные атрофии и т.д.) и снижается при нарастании тяжести заболевания. Буниной была отмечена высокая вариабельность содержания и распределения между ядром и цитоплазмой РНК в двигательных нейронах передних рогов спинного мозга и ядер черепно-мозговых нервов [22]. Также удалось выявить большую роль биохимического обмена нуклеиновых кислот, определена и описана связь нуклеиновых кислот и внутриклеточных включений, образующихся при развитии заболевания. Цитоплазматические, оксифильные включения были обнаружены во всех случаях БАС в ганглиозных клетках передних рогов спинного мозга [29]. Эти включения являются типичным патоморфологическим признакам БАС. Бунина описала их для нескольких случаев семейной формы БАС, однако, в дальнейшем они были описаны и для спорадической формы БАС [30]. Примечательно, долгое время ошибочно считалось, что впервые их описал японский невропатолог Асао Хирано (Asao Hirano), однако он сделал свое описание в 1965 г., а Бунина в 1962 г [31,32].
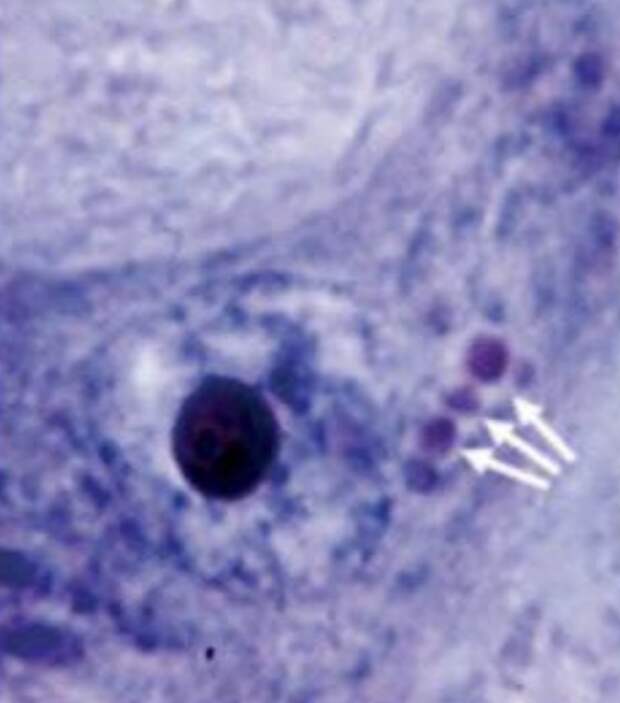
Тельца Буниной
Продолжение эксперимента на обезьянах
Предварительные результаты эксперимента по инфицированию животных ликвором от больных БАС пациентов вдохновили группу Зильбера на продолжение исследований в данном направлении дальнейшие. Было принято решение увеличить количество животных и, чтобы исключить воздействия каких-либо сторонних факторов, как это было в первом эксперименте (животные, задействованные в первом эксперименте, ранее были использованы как модельные в изучении опухолеобразования, а автор объясняет это тем, что иных животных не было в наличии в питомнике), в дальнейшем заражать обезьян уже ликвором, полученным от первых трех макак, достоверно заболевших БАС.
Они, в свою очередь, были инокулированы суспензией продолговатого и спинного мозга, полученной от трех больных различными формами БАС. Схема эксперимента выглядит следующим образом: всего, материалом, полученным от пациентов с БАС, были инокулированы 21 обезьяна, две вскоре погибли от других инфекций, из 19 оставшихся у 9 животных развились симптомы БАС. У трех животных наблюдалась развернутая картина заболевания, практически полностью идентичная с таковой у больных БАС. Симптомы нейродегенерации у обезьян, инокулированных материалом от двух больных с различными формами БАС (бульбарная и пояснично – крестцовая), также были аналогичны таковым у пациентов с БАС. В дальнейшем у обезьян с симптоматикой БАС был произведен забор ликвора, который был использован для заражения следующих поколений Macacus rhesus. От двух больных обезьян был получен материал, которым заразили 17 макак, из них заболели 7, далее от одной из этих 7 больных заразили еще 5. Все последующие заражения дали результат в виде развития заболевания у подопытных животных (Рис.1) [22]
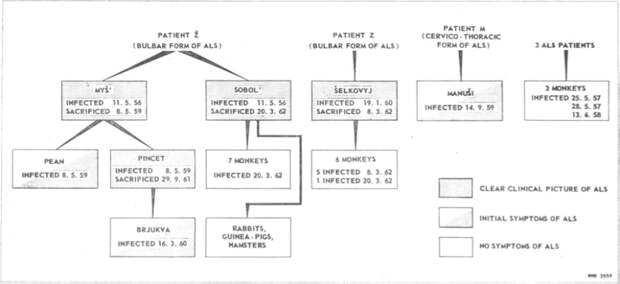
Схема эксперимента Буниной
В 1963 году коллектив авторов во главе с Зильбером публикует англоязычную статью, и, начиная с этого момента исследования, его группы стали доступны мировому научному сообществу [26]
Попытки повторить советский эксперимент на западе
Работа по иммунологической характеристике БАС продолжилась, однако сотни попыток высеять, охарактеризовать и верифицировать вирусный агент оказываются бесплодными. Примечательно, что исследователи проводили параллели с такими заболеваниями как Куру, Висна-маеди и синдром Крейтцфельдта-Якоба (сегодня известные как трансмиссивные прионные заболевания), предполагая, что введенный инфицирующий агент в виде экстракта мозга больных людей не инфицировал обезьян, а лишь активировал латентную, уже имеющуюся у животных инфекцию [23]. Фраза «латентная инфекция» прекрасно ложится в контекст молекулярного патогенеза прионных заболеваний, например таких как Куру. Результаты проведенных экспериментов указывали на то, что предположительное инфекционное начало достоверно находится в ликворе, которым заражали обезьян. В 1973 г. Фредерик Вольфргамм и Лоуренс Маерс из Центра неврологических исследований Рида в Калифорнии продемонстрировали токсичность сыворотки крови, полученной от пациентов с БАС, при обработке ею культуры клеток передних рогов спинного мозга мыши. Из 37 протестированных сывороток, полученных от пациентов с различными формами БАС, 27 сывороток (70%) селективно разрушали клетки передних рогов спинного мозга. Причем цитотоксическому воздействию были подвержены только нейроны. Токсический фактор в сыворотке не диализировался – это, предположительно, говорило о том, что данным фактором является молекула белковой природы.
Более того, 44 контрольные сыворотки, полученные от пациентов, страдающих другими нейродегенеративными заболеваниями, не продемонстрировали никакого токсического эффекта на нейроны [33]. Успешная трансмиссия заболевания Крейтцфельдта-Якоба от больных пациентов семи обезьянам в 1969 г. выполненная группой исследователей под руководством Гайдузека окончательно вселила уверенность, что заболевание передается «особым» вирусом [34]. Это обстоятельство позволило исследователям включиться в работу по изучению этиологии БАС. Исследовательская группа попыталась перенести заболевание от 22 индейцев чаморро с Западно-Тихоокеанской (Гуам-тип) формой БАС на ряд мелких лабораторных животных, таких как, крысы и мыши и на шимпанзе. Однако авторы не получили каких-либо значимых результатов, даже несмотря на то, что пытались заразить животных материалом второго пассажа от Macacus rhesus, полученного от советских исследователей [22,35]
Необходимо отметить, что на сегодняшний день Западно-Тихоокеанскую форму заболевания выделяют отдельно наряду со спорадической и семейной формами БАС. Гуам-тип клинически повторяет БАС, однако, у пациентов практически всегда присутствует паркинсонический синдром. Этот тип наблюдаются в трех эндемичных регионах: Марианские острова, преимущественно остров Гуам, полуостров Кии в Японии и западное побережье Новой Гвинеи. Предполагалось, что заболевание носит наследственный характер, т. к. частота распространения была очень высокой и затрагивала почти все возрастные группы в популяции. Заболевание впервые было описано как «наследственный паралич» [36]. Высказывалось множество предположений относительно этиологии Западно-Тихоокеанской формы БАС, однако выяснилось и позднее подтвердилось предположение об экзогенной природе данного типа нейродегенерации. Для Гуам-типа БАС была достоверно доказана роль экзогенных растительных токсинов семян сагового дерева, которые активно использовались в пищу индейцами Чаморро на острове. Из семян было выделено два нейротоксина: β-N-метиламино-L-аланин и циказин [30]. Заболеваемость данной формой БАС резко снизилась с приходом в регион США [36], что можно обяснить сменой рациона у населения и увеличением уровня жизни в целом. По сей день причины данной эндемичной формы заболевания до конца не ясны, предполагается, что оно развивается на фоне генетических нарушений, в то время как факторы внешней среды могут модифицировать клинические фенотипы БАС [37–39]
Все советские исследования по переносу заболевания от человека к животному базировались на спорадических формах БАС, что заставляет задуматься: правильно ли были выбраны объекты в работе Гайдузека, и, возможно, именно различие в форме БАС стало решающим в неудачной попытке повторить результаты наших соотечественников?
Американские авторы в своих неудачных попытках переноса заболевания БАС от человека к примату акцентируют внимание на том, что ряд исследователей не обнаружили ни клинической, ни патологической картины БАС у Macacus rhesus, инфицированных в советской лаборатории. Кроме того, Хирано в 1965 г. сообщил, что не смог обнаружить патогистологических изменений в тканях спинного и головного мозга обезьян (он исследовал гистологические срезы тканей головного и спинного мозга животных, приготовленные и переданные ему Буниной), присущих БАС. Это обстоятельство представляется весьма сомнительным, речь идет о научной репутации исследовательской группы Зильбера, и, прежде, чем выносить результаты на суд мировой научной общественности, Буниной были приготовлены не одна сотня срезов тканей головного и спинного мозга обезьян с развившимся БАС-подобным синдромом [22]. При помощи этих срезов были описаны патогистологические признаки, присущие БАС, более того обнаружены и охарактеризованы оксифильные тельца – сегодня носящие имя исследователя, описавшего их; данные включения являются типичным патоморфологическим признакам БАС. После нескольких неудачных попыток повторить советский эксперимент и получить релевантные результаты американская исследовательская группа сделала вывод, что невозможно никак связать неврологические симптомы, напоминающие таковые при прогрессии БАС с конкретным инфицирующим агентом, иначе говоря, ликвор, которым иммунизировали обезьян, не может считаться причиной развития БАС-подобного синдрома у Macacus rhesus и в нем же не может быть этиологического начала для развития заболевания у человека [35].
Современные представления о природе БАС
Сегодня утверждать, что БАС имеет вирусную природу, в общем и целом, неверно. В некоторых случаях заболевания рядом исследователей были выявлены определённые последовательности энтеровирусной РНК в клетках передних рогов спинного мозга больных [40–42] и была предложена гипотеза, о том, что причиной спорадических форм БАС может быть персистирующуя энтеровирусная инфекция, которая впоследствии не нашла убедительных доказательств [43,44]. К настоящему моменту медико-генетические исследования выявили в дополнение к уже известному гену SOD1 ряд других генов, мутации в которых приводят к образованию патогенных форм, кодируемых ими белков, и к развитию нейродегенеративного процесса с поражением двигательных нейронов. Известно более 100 генов ассоциированых с БАС, и множество мутаций находят в генах РНК-связывающих белков (TDP-43, FUS, hnRNPA1, hnRNPA2/B1, MATR3, ATXN2, TAF15, TIA-1 и EWSR1) [10,11].
К сожалению, причины развития спорадических форм БАС, до сих пор не ясны. Это многофакторное заболевание со сложным патогенезом (Рис. 2), где клетки микроглии могут активировать нейровоспалительные реакции путем синтеза цитокинов, астроциты задействованы в повреждении двигательных нейронов путем высвобождения медиаторов воспаления, таких, как оксид азота, простагландин Е2 и др. Накопление кислородных радикалов и развитие окислительного стресса, абберантный процессинг РНК, неправильное сворачивание белков, их агрегация и переход в нерастворимую форму так же могут вызвать гибель нейрона, а нарушение аксонального транспорта, дисфункции митохондрий и д.р. способны привести к апоптозу клетки [45].
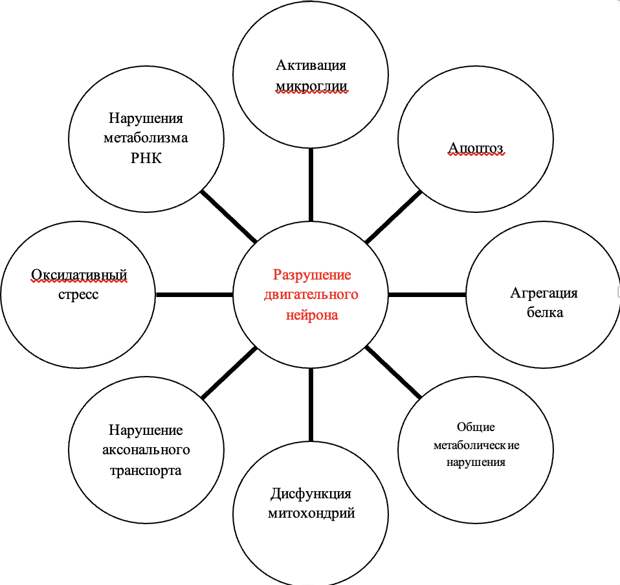
Вклад различных путей в формирование БАС
Белки, содержащие прионоподобные домены, участвуют в патогенезе БАС
Все без исключения формы БАС характеризуются наличием патогенетических включений, образованных агрегированными формами ряда ассоциированных с этим заболеванием белков, что классифицирует БАС как протеинопатию [9]. Примечательно, что в патогенезе БАС задействованы белки, такие как FUS и ТDP-43, имеющие в своем составе прионовые мотивы. Они являются РНК/ДНК-связывающими белками, способными к агрегации. В дополнение к их участию в развитии БАС, белки имеют некоторые, ключевые структурные особенности. С помощью биоинформатических методов, исследователи недавно обнаружили прионоподобные домены в белках FUS и ТDP-43. Как и в прионах дрожжей, эти домены обогащены незаряженными полярными аминокислотами (аспарагин, глютамин и тирозин) и глицином. В результате проверки на наличие прионоподобных последовательностей, 27 879 белков из протеома человека, FUS и TDP-43, занимают 15 и 69-е место, соответственно. Прионоподобный домен в белке FUS находится на N-конце, тогда как в белке TDP-43 – на С-конце белка. Было показано, что и некоторые другие РНК-связывающие белки также содержат прионоподобные мотивы, и, возможно, они так же склонны к агрегации. Все это говорит в пользу того, что, возможно, такие белки причастны к развитию БАС [46].
Некоторые авторы пытаются объяснить распространение заболевания в рамках одного организма, исходя из того, что данные белки обладают прионоподобыми свойствами и способны инициировать агрегацию. Хотя и нет точных данных на этот счет, однако, ясно, что картина распространения заболевания в пределах нервной системы одного пациента весьма схожа при БАС [47–49].
Для мутантных вариантов белка SOD1 была показана возможность вызывать агрегацию у нативных аналогов. Белки TDP-43 и FUS являются компонентами стресс-гранул и в трансфецированных клетках ко-локализуется с ними, а FUS-позитивные включениях в нейронах пациентов с БАС и ФТЛД содержат молекулярные маркеры стресс-гранул. Стресс-гранулы и P-тельца – это временные цитоплазматические структуры, содержащие РНК, РНК связывающие белки, инициаторы трансляций и polyA-связывающий белок (PABP). Они необходимы для секвестрации мРНК во время клеточного стресса для того, чтобы ингибировать инициацию трансляции [46]. Образование стресс гранул происходит за счет последовательной агрегации нескольких РНК-связывающих белков в комплексе с молекулами РНК. Эта физиологическая реакция на клеточный стресс может быть первоначальным пусковым механизмом для формирования патогенных включений, так как увеличение локальной концентрации белков и РНК, могут способствовать агрегации TDP-43 и/или FUS (Рис.3, А). Неправильное сворачивание и, как следствие агрегация белка SOD1 инициирует seeding-реакцию в культуре клеток. Различные стрессовые факторы могут инициировать неправильную сборку стресс гранул, что ведет к формированию конформационных изменений основных белков, входящих в состав стресс гранул — TDP-43 и FUS (Рис.3, В). В клеточных культурах для белка SOD1 была показана возможность «передачи информации о неправильной сборке», т. е. наблюдалась типичная прионогенная картина распространения заболевания. Для белков TDP — 43 и FUS еще не обнаружена такая возможность распространения от клетки к клетке in vivo, или в животных моделях, как это было показано, к примеру, для болезни Альцгеймера [50].
Ранее было показано, что в клеточных моделях с экспрессией транкированных форм белков, для развития токсичности TDP-43 необходимы С-концевая часть и РРД (РНК-распознающий домен) белка, но для конструктов белка FUS, содержащих N-концевую часть с прионоподобными мотивами и РРМ (РНК-распознающий мотив) (1-373) нормальной оказалась полная ядерная локализация, а формы белка были нетоксичными. Однако для придания белку свойств токсичности и склонности к цитоплазматической агрегации достаточно было добавить к конструкту первый аргинин-глицин-глицин богатый мотив, хотя он и не был столь токсичным как полноразмерный белок. Примечательно, что первый аргинин-глицин-глицин богатый мотив белка FUS также содержит участок, который напоминает прионовый домен дрожжей, и возможно два прионо-подобных мотива в белке FUS необходимы для активации его агрегационных свойств. А делетирование N-концевого, прионо-подобного домена белка FUS полностью предотвращает его агрегацию. Таким образом, в отличие от белка TDP-43, для агрегации токсичности которого необходим С-концевой участок прионоподобного домена, а также части аргинин-глицин-глицин богатого участка, для белка FUS необходимы N- концевой прионо-подобный домен, РРД и первый аргини-глицин-глицин богатый мотив (Рис.3, С) [46]. Интересно, что мутации в прионоподобных доменах белков FUS и TDP-43 способны усилить их склонность к агрегации, а мутации в области сигнала ядерной локализации – эксклюзию из белков из ядра [51]. Nonaka T. с соавторами показал, что агрегированные формы белка TDP-43 имеют свойства, присущие прионам. В работе исследователи обрабатывали культуру клеток SH-SY5Y агрегированными белками TDP-43, выделенными из мозга больных БАС и ФТЛД. Такие нерастворимые, агрегированные белки вызывали агрегацию нативных TDP-43. Причем обработка протеазами, формалином не влияла на способность активировать агрегацию нативного TDP-43 в клеточной культуре [52].
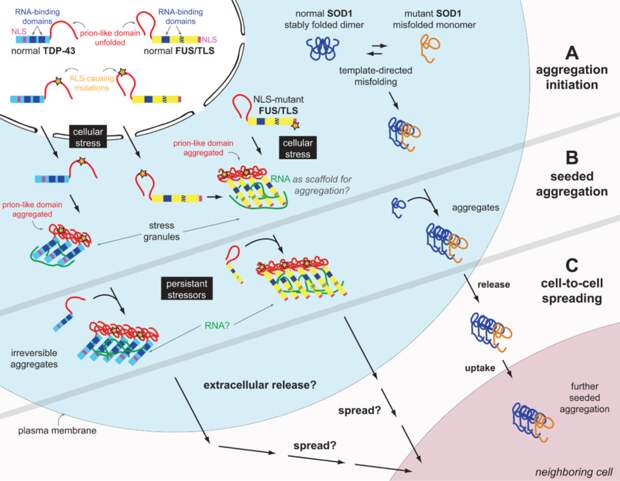
В свете полученных на сегодняшний день данных, о гипотетически возможном прионоподобном механизме развития БАС, некоторые авторы считают, что БАС может быть, прежде всего, болезнью коры головного мозга. Некоторые авторы предполагают, что, изначально формирование TDP-43-позитивных включений происходит в пирамидальных клетках коры головного мозга, а все остальные повреждения, возникающие в подкорковых областях вторичны, и распространяются по нисходящим аксонным путям [49]. И такой механизм может быть объяснен только с точки зрения свойств, присущих прионам, или seeding prion-like белкам. Однако пока, что все это только гипотезы. Основательных данных о том, как будут себя вести, к примеру, белки TDP-43 и FUS, in vivo в сложных модельных системах млекопитающих, сегодняшний день еще нет.
Возвращаясь к советским исследованиям
Сегодня сложно судить о правдивости и значимости эксперимента группы Зильбера на обезьянах. Вполне вероятно, американской группе исследователей не удалось повторить эксперимент в силу некоторых причин, оной из которых может быть неудачный выбор формы БАС для переноса его в организм приматов. И возможно, что советские исследователи уже тогда вплотную подошли к проблеме, прогресс в изучении, которой мы видим сегодня, но были не поняты коллегами. Удивительно, но Бунина указывает на важную роль РНК в развитии патологического процесса. Как уже известно – метаболизм РНК играет существенную роль в развитии БАС. И что примечательно, автором указывается абнормальное распределение РНК в клетках передних рогов спинного мозга, высокая вариабельность ее распределения. Это косвенно может указывать на то, что Бунина и ее коллеги могли столкнуться с БАС, вызванным нарушениями структурной функциональной активности белков TDP-43 и FUS/TLS, нормальная работа, которых чрезвычайно важна для функционирования клетки двигательного нейрона. К тому же важно, что в эксперименте были использованы страдающие БАС пациенты только со спорадической формой заболевания. Гайдузек предположил, что советские коллеги включили в исследование животных, уже страдающих различными заболеваниями, на фоне которых, возможно могли развиться симптомы, описанные советскими исследователями. Однако Бунина в своих работах подчеркивает, что с Сухумском питомнике, где содержались и разводились животные, многолетний опыт наблюдения за обезьянами не выявил никаких явных и латентных заболеваний, а если бы данные заболевания протекали в скрытой форме, то они бы их рано или поздно обнаружили (эксперимент продолжался более 10 лет).
Заключение
Никто не застрахован от ошибок, тем более в научной среде. Ученные часто ошибаются: строят ложные гипотезы, проводят неинформативные эксперименты, некорректно, или неаккуратно ведут наблюдения, ошибаются в статистическом анализе, делают ложные выводы. В научном сообществе обязательно укажут на ошибку (нет – если ученный, совершивший ее намерено, гениальный фальсификатор), поправят, будь то редактор научного журнала научный руководитель или кто-то из этой же области знаний. Исследователь хотел видеть желаемое в действительном, да так, что не заметил откровенной «фикции» в своих данных, или же наоборот специально все подвел к «нужному» для себя, возможно, и мировому научному сообществу результату. В этом случаем исход предрешен: исследователь начнет все сначала. А как быть, если изыскания исследователь проводил верно, и не один десяток лет! Но вот коллеги вовсе не спешат признавать его достижений и выводов, еще и понукают: развели фикцию, цифры подогнали и т.д. А выхода два. Первый – драться до последнего, доказывая всем свою правоту, или отступить, уйти и оставить последнее слово за разоблачителями. Именно второй вариант выбрала для себя выдающаяся ученый, невролог, возможно, «докопавшийся» до истины в вопросе этиологии бокового амиотрофического склероза Татьяна Львовна Бунина.
Текст: Евгений Броновицкий
1. Сергей Иллариошкин: «По самым скромным подсчетам, около 35 млн людей в мире страдают болезнью Альцгеймера и болезнью Паркинсона» [Electronic resource]. 2020. URL: accessed: 14.12.2022.
2. В России создадут регистр пациентов с болезнью Альцгеймера [Electronic resource]. 2022. URL: accessed: 14.12.2022.
3. Новые подходы к лечению болезни Альцгеймера обсудят на OpenBio-2022 [Electronic resource]. 2022. URL: Новые подходы к лечению болезни Альцгеймера обсудят на OpenBio-2022 (accessed: 14.12.2022).
4. Инновационное российское лекарство замедлит наступление деменции [Electronic resource]. 2022. URL: accessed: 14.12.2022.
5. Новое исследование ученых закладывает основу корректировки нейрологических болезней от эпилепсии до Альцгеймера [Electronic resource]. 2022. URL: accessed: 14.12.2022.
6. Sayed N. et al. Gene therapy: Comprehensive overview and therapeutic applications // Life Sci. 2022. Vol. 294. P. 120375.
7. Fang T. et al. Gene Therapy in Amyotrophic Lateral Sclerosis // Cells. 2022. Vol. 11, № 13. P. 2066.
8. Amado D.A., Davidson B.L. Gene therapy for ALS: A review // Molecular Therapy. 2021. Vol. 29, № 12. P. 3345–3358.
9. Shelkovnikova T.A. et al. Proteinopathies — forms of neurodegenerative disorders with protein aggregation-based pathology. // Molekuliarnaia biologiia. 2012. Vol. 46, № 3. P. 402–415.
10. Xue Y.C. et al. Dysregulation of RNA-Binding Proteins in Amyotrophic Lateral Sclerosis. // Front Mol Neurosci. 2020. Vol. 13. P. 78.
11. Kapeli K., Martinez F.J., Yeo G.W. Genetic mutations in RNA-binding proteins and their roles in ALS. // Hum Genet. 2017. Vol. 136, № 9. P. 1193–1214.
12. Shelkovnikova T.A. Modelling FUSopathies: focus on protein aggregation. // Biochem Soc Trans. 2013. Vol. 41, № 6. P. 1613–1617.
13. The complementary structure of deoxyribonucleic acid // Proc R Soc Lond A Math Phys Sci. The Royal Society London , 1954. Vol. 223, № 1152. P. 80–96.
14. The Genetic Code // Cold Spring Harb Symp Quant Biol. 1966. Vol. 31, № 0. P. 1–1.
15. Sim V.L. Prion Diseases // Viral Infections of the Human Nervous System. Basel: Springer Basel, 2013. P. 371–401.
16. Gajdusek D.C. Fantasy of a “Virus” from the Inorganic World: Pathogenesis of Cerebral Amyloidoses by Polymer Nucleating Agents and/or “Viruses.” 1989. P. 481–499.
17. Greig J.R. Scrapie in sheep // Journal of Comparative Pathology and Therapeutics. 1950. Vol. 60. P. 263–266.
18. Гардашьян A.M. Вирус болезни скрепи // Успехи современной биологии. 1967. Vol. 3. P. 429–437.
19. CRICK F. Central Dogma of Molecular Biology // Nature. 1970. Vol. 227, № 5258. P. 561–563.
20. ter Meulen V., Hall W.W. Slow Virus Infections of the Nervous System: Virological, Immunological and Pathogenetic Considerations // Journal of General Virology. 1978. Vol. 41, № 1. P. 1–25.
21. Gajdusek D.C., Zigas V. Degenerative Disease of the Central Nervous System in New Guinea // New England Journal of Medicine. 1957. Vol. 257, № 20. P. 974–978.
22. Бунина Т. Л. Боковой амиотрофический склероз. (Клинико-морфологическое и экспериментальное исследование): Дисс. канд. мед. наук. Москва: Ин-т неврологии Акад. мед. наук СССР, 1963.
23. Бунина Т. Л. Патогенез бокового амиотрофического склероза (Гистохимические, клинико-биохимические и экспериментальные исследования): Диссертация д-ра мед. наук. Москва: Ин-т неврологии Акад. мед. наукСССР, 1975.
24. GAJDUSEK D.C., GIBBS C.J., ALPERS M. Experimental Transmission of a Kuru-like Syndrome to Chimpanzees // Nature. 1966. Vol. 209, № 5025. P. 794–796.
25. Gajdusek D.C. et al. TRANSMISSION EXPERIMENTS WITH KURU IN CHIMPANZEES AND THE ISOLATION OF LATENT VIRUSES FROM THE EXPLANTED TISSUES OF AFFECTED ANIMALS // Ann N Y Acad Sci. 1969. Vol. 162, № 1 Experimental. P. 529–550.
26. ZILBER L.A. et al. STUDY OF THE ETIOLOGY OF AMYOTROPHIC LATERAL SCLEROSIS. // Bull World Health Organ. 1963. Vol. 29, № 4. P. 449–456.
27. ZIL’BER L.A. et al. On the possible viral etiology of lateral amyotrophic sclerosis. // Vopr Virusol. 1962. Vol. 7. P. 520–528.
28. Гардашьян А.М. Серологические исследования при изучении БАС // Биллютень экспериментальной биологии и медицины. 1970. Vol. 4. P. 88–90.
29. BUNINA T.L. On intracellular inclusions in familial amyotrophic lateral sclerosis // Zh Nevropatol Psikhiatr Im S S Korsakova. 1962. Vol. 62. P. 1293–1299.
30. БОЙКО А.Н. et al. Боковой амиотрофический склероз / ed. ЗАВАЛИШИН И.А. Москва: ГЭОТАР-Медиа, 2009.
31. Rowland L.P. T.L. Bunina, Asao Hirano, and the post mortem cellular diagnosis of amyotrophic lateral sclerosis // Amyotrophic Lateral Sclerosis. 2009. Vol. 10, № 2. P. 74–78.
32. Okamoto K., Mizuno Y., Fujita Y. Bunina bodies in amyotrophic lateral sclerosis // Neuropathology. 2008. Vol. 28, № 2. P. 109–115.
33. Wolfgram F., Myers L. Amyotrophic Lateral Sclerosis: Effect of Serum on Anterior Horn Cells in Tissue Culture // Science (1979). 1973. Vol. 179, № 4073. P. 579–580.
34. Gibbs C.J., Gajdusek D.C. Infection as the Etiology of Spongiform Encephalopathy (Creutzfeldt-Jakob Disease) // Science (1979). 1969. Vol. 165, № 3897. P. 1023–1025.
35. Gibbs C.J., Gajdusek D.C. Amyotrophic lateral sclerosis, Parkinson’s disease, and the amyotrophic lateral sclerosis-Parkinsonism-dementia complex on Guam: a review and summary of attempts to demonstrate infection as the aetiology. // J Clin Pathol Suppl (R Coll Pathol). 1972. Vol. 6. P. 132–140.
36. Haddock R.L., Chen K.-M. Amyotrophic lateral sclerosis and diabetes on Guam: changing patterns of chronic disease in an island community. // Southeast Asian J Trop Med Public Health. 2003. Vol. 34, № 3. P. 659–661.
37. Mitchell J.D. Heavy metals and trace elements in amyotrophic lateral sclerosis. // Neurol Clin. 1987. Vol. 5, № 1. P. 43–60.
38. Chen K.M. Disappearance of ALS from Guam: implications for exogenous causes. // Rinsho Shinkeigaku. 1995. Vol. 35, № 12. P. 1549–1553.
39. Muddasir Qureshi M. et al. Analysis of factors that modify susceptibility and rate of progression in amyotrophic lateral sclerosis (ALS) // Amyotrophic Lateral Sclerosis. 2006. Vol. 7, № 3. P. 173–182.
40. Pamphlett R., Lina B., Karpati G. Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS // Neurology. 2000. Vol. 55, № 9. P. 1420–1421.
41. Woodall C., Graham D. Evidence for neuronal localisation of enteroviral sequences in motor neurone disease/amyotrophic lateral sclerosis by in situ hybridization // European Journal of Histochemistry. 2009. Vol. 48, № 2. P. 129.
42. Berger M.M. et al. Detection and cellular localization of enterovirus RNA sequences in spinal cord of patients with ALS // Neurology. 2000. Vol. 54, № 1. P. 20–20.
43. Swanson N.R., Fox S.A., Mastaglia Frank.L. Search for persistent infection with poliovirus or other enteroviruses in amyotrophic lateral sclerosis-motor neurone disease // Neuromuscular Disorders. 1995. Vol. 5, № 6. P. 457–465.
44. Nix W.A. et al. Failure to detect enterovirus in the spinal cord of ALS patients using a sensitive RT-PCR method // Neurology. 2004. Vol. 62, № 8. P. 1372–1377.
45. Chen S. et al. Genetics of amyotrophic lateral sclerosis: an update // Mol Neurodegener. 2013. Vol. 8, № 1. P. 28.
46. Gitler A.D., Shorter J. RNA-binding proteins with prion-like domains in ALS and FTLD-U // Prion. 2011. Vol. 5, № 3. P. 179–187.
47. Talbot K. Amyotrophic lateral sclerosis: cell vulnerability or system vulnerability? // J Anat. 2014. Vol. 224, № 1. P. 45–51.
48. Kanouchi T., Ohkubo T., Yokota T. Can regional spreading of amyotrophic lateral sclerosis motor symptoms be explained by prion-like propagation? // J Neurol Neurosurg Psychiatry. 2012. Vol. 83, № 7. P. 739–745.
49. Braak H. et al. Amyotrophic lateral sclerosis—a model of corticofugal axonal spread // Nat Rev Neurol. 2013. Vol. 9, № 12. P. 708–714.
50. Fernández-Borges N. et al. Infectivity versus Seeding in Neurodegenerative Diseases Sharing a Prion-Like Mechanism // Int J Cell Biol. 2013. Vol. 2013. P. 1–9.
51. Polymenidou M., Cleveland D.W. The Seeds of Neurodegeneration: Prion-like Spreading in ALS // Cell. 2011. Vol. 147, № 3. P. 498–508.
52. Nonaka T. et al. Prion-like Properties of Pathological TDP-43 Aggregates from Diseased Brains // Cell Rep. 2013. Vol. 4, № 1. P. 124–134.
Свежие комментарии